DIRAC Spatial Multi-Omics — Horizontal Integration¶
Horizontal annotation with DIRAC. This Quickstart explains how the demo runs and what each step does, for both single-modality (RNA→RNA) and dual-modality (RNA+ATAC→RNA+ATAC) annotation using spatial graphs, plus an optional novel cell type discovery stage.
Table of Contents¶
Overview & Scope
Environment & Data
Load Packages
Single-Modality Annotation (RNA → RNA)
Metrics & Visualization
Dual-Modality Annotation (RNA+ATAC → RNA+ATAC)
Novel Cell Type Discovery (Confidence Filtering)
Tips & Troubleshooting
1. Overview & Scope¶
Goal: Transfer cell type labels from a source dataset to a target dataset (horizontal integration) using DIRAC with spatial graphs.
Included in this demo:
Single-modality annotation: RNA → RNA
Dual-modality annotation: RNA+ATAC → RNA+ATAC (feature concatenation with explicit modality splits)
Optional novel-cell discovery: apply confidence filtering (e.g., threshold = 0.9) so low-confidence predictions are marked “unassigned”, revealing putative missing/unknown cell types when the reference is incomplete.
High-level flow
Load AnnData for source and target.
Preprocess each dataset (normalize → log1p → scale).
Build spatial graphs: multi-batch (source) and single-sample (target).
Initialize DIRAC’s annotate_app.
Package data via
_get_data(...)(optionally split into subgraphs withnum_parts_*).Build a GNN via
_get_model(...)(e.g., GraphSAGE or GAT).Train
_train_dirac_annotate(...)to learn features & predict labels on the target.(Optional) Enable confidence filtering during training to tag low-confidence predictions as
"unassigned"; tuneconfidence_threshold(e.g., 0.8–0.95) based on data quality and reference completeness.Write back embeddings/predictions; evaluate accuracy/precision/recall/F1 on assigned cells, report Unassigned Rate, and visualize spatial maps and confidence.
2. Environment & Data¶
Dependencies
Python ≥ 3.9
scanpy, anndata, numpy, matplotlib, scikit-learn, torch
DIRAC codebase available. If not installed, follow: https://dirac-tutorial.readthedocs.io/en/latest/install.html
Paths used in the demo
data_path:
../DIRAC-main/data/scMultiSimsave_path:
./Resultsmethods tag:
"DIRAC"
Expected input files
Single-modality:
source_RNA.h5ad,target_RNA.h5adDual-modality: additionally
source_ATAC.h5ad,target_ATAC.h5ad
Required fields in the AnnData
obsm["spatial"]for spatial coordinates (source & target)obs["batch"]for source (used by multi-batch graph building)obs["cell.type"]for source labels and target ground truth (for metrics)
What “mask = 0.3” means (important)
A dropout mask is applied independently to both RNA and ATAC matrices.
Roughly 30% of entries are randomly set to zero (element-wise), i.e., random sparsification at rate 0.3 in each modality.
3. Load Packages¶
[1]:
import os
import scanpy as sc
import numpy as np
import matplotlib.pyplot as plt
import anndata
import sklearn
import sodirac as sd
sd.utils.seed_torch(seed=0)
data_path = "../DIRAC-main/data/scMultiSim"
save_path = './Results'
methods = "DIRAC"
4. Single-Modality Annotation (RNA → RNA)¶
What this part does
Uses RNA only. Learns from
source_RNA.h5ad(with cell types) and predicts labels fortarget_RNA.h5ad.Builds a multi-batch spatial graph for source (accounts for
obs["batch"]) and a single-sample spatial graph for target.
Step-by-step (as in your script)
[2]:
# 1) Load source/target
source_RNA = anndata.read_h5ad(os.path.join(data_path, "source_RNA.h5ad"))
target_RNA = anndata.read_h5ad(os.path.join(data_path, "target_RNA.h5ad"))
print(source_RNA); print(target_RNA)
# 2) Preprocess (normalize → log1p → scale)
sc.pp.normalize_total(source_RNA)
sc.pp.log1p(source_RNA)
sc.pp.scale(source_RNA)
sc.pp.normalize_total(target_RNA)
sc.pp.log1p(target_RNA)
sc.pp.scale(target_RNA)
# 3) Spatial graphs
# - Source: multi-batch kNN graph (uses spatial + batch)
source_edge_index = sd.utils.get_multi_edge_index(source_RNA.obsm["spatial"], source_RNA.obs["batch"], n_neighbors=10)
# - Target: single-sample kNN graph
target_edge_index = sd.utils.get_single_edge_index(target_RNA.obsm["spatial"], n_neighbors=10)
# 4) Initialize DIRAC annotation app
annotate = sd.main.annotate_app(save_path=save_path, use_gpu=True)
# 5) Package data for training
samples = annotate._get_data(
source_data=source_RNA.X,
source_label=source_RNA.obs["cell.type"],
source_edge_index=source_edge_index,
target_data=target_RNA.X,
target_edge_index=target_edge_index,
num_parts_source=source_RNA.shape[0] // 200,
num_parts_target=target_RNA.shape[0] // 200,
)
# 6) Build model (GraphSAGE in this demo)
models = annotate._get_model(samples=samples)
# 7) Train
results = annotate._train_dirac_annotate(samples=samples, models=models, n_epochs=100)
# np.savez(os.path.join(save_path, "DIRAC_results.npz"), **results)
# 8) Write back embeddings & predictions
source_RNA.obsm[f"{methods}_embed"] = results["source_feat"]
target_RNA.obsm[f"{methods}_embed"] = results["target_feat"]
target_RNA.obs[f"{methods}_pred"] = results["target_pred"]
target_RNA.obs[f"{methods}"] = target_RNA.obs[f"{methods}_pred"].map(results["pairs"])
AnnData object with n_obs × n_vars = 11971 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 3029 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
Average neighbors per node (directed): 10.00 (edges=119710, nodes=11971)
Average neighbors per node (directed): 10.00 (edges=30290, nodes=3029)
Identified 2 unique domains.
Computing METIS partitioning...
Done!
Computing METIS partitioning...
Done!
DIRAC annotate training..: 100%|█| 10
5. Metrics & Visualization¶
Metrics (weighted, as in your script)
Accuracy, Precision (weighted), Recall (weighted), F1 (weighted) using sklearn.
Spatial visualization
Compare ground truth vs predicted labels on the target:
[3]:
metrics_all = {
"Accuracy Score":
float(sklearn.metrics.accuracy_score(target_RNA.obs["cell.type"], target_RNA.obs[f"{methods}"])),
"Precision Score":
float(sklearn.metrics.precision_score(target_RNA.obs["cell.type"], target_RNA.obs[f"{methods}"], average='weighted')),
"Recall Score":
float(sklearn.metrics.recall_score(target_RNA.obs["cell.type"], target_RNA.obs[f"{methods}"], average='weighted')),
"F1 Score":
float(sklearn.metrics.f1_score(target_RNA.obs["cell.type"], target_RNA.obs[f"{methods}"], average='weighted'))}
print(metrics_all)
sc.pl.embedding(target_RNA, basis='spatial', color=['cell.type', f'{methods}'], title=['Ground truth',f"{methods} ACC {metrics_all['Accuracy Score']}"], s=50, show=False)
{'Accuracy Score': 0.880488610102344, 'Precision Score': 0.8821684104621875, 'Recall Score': 0.880488610102344, 'F1 Score': 0.8804397263487906}
[3]:
[<Axes: title={'center': 'Ground truth'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'DIRAC ACC 0.880488610102344'}, xlabel='spatial1', ylabel='spatial2'>]

6. Dual-Modality Annotation (RNA+ATAC → RNA+ATAC)¶
What this part does
Concatenates RNA and ATAC features per cell and performs label transfer across modalities.
Uses
combine_multimodal_adatas(...)to build a single AnnData with stacked features.Provides a
split_listto tell DIRAC where each modality lives in the concatenated matrix.
Subgraph-settings(num_part)
The spatial graph is split into
num_parts_*subgraphs. More parts → smaller subgraphs → lower peak memory but more overhead.Ensure at least 1 part:
max(1, n_cells // 200).When to increase: GPU OOM or very large tissues.
Notes on ``split_list``
It must enumerate non-overlapping half-open ranges
(start, end)that exactly cover the concatenated feature columns.Make sure the ranges line up with the same order you used in
combine_multimodal_adatas({...}).
Step-by-step
[4]:
# 1) Load inputs
# RNA
source_adata_RNA = anndata.read_h5ad(os.path.join(data_path, "source_RNA.h5ad"))
target_adata_RNA = anndata.read_h5ad(os.path.join(data_path, "target_RNA.h5ad"))
print(source_adata_RNA); print(target_adata_RNA)
# ATAC
source_adata_ATAC = anndata.read_h5ad(os.path.join(data_path, "source_ATAC.h5ad"))
target_adata_ATAC = anndata.read_h5ad(os.path.join(data_path, "target_ATAC.h5ad"))
print(source_adata_ATAC); print(target_adata_ATAC)
# 2) Preprocess each modality
sc.pp.normalize_total(source_adata_RNA); sc.pp.log1p(source_adata_RNA); sc.pp.scale(source_adata_RNA)
sc.pp.normalize_total(target_adata_RNA); sc.pp.log1p(target_adata_RNA); sc.pp.scale(target_adata_RNA)
sc.pp.normalize_total(source_adata_ATAC); sc.pp.log1p(source_adata_ATAC); sc.pp.scale(source_adata_ATAC)
sc.pp.normalize_total(target_adata_ATAC); sc.pp.log1p(target_adata_ATAC); sc.pp.scale(target_adata_ATAC)
# 3) Concatenate modalities (feature-wise)
source_adata = sd.utils.combine_multimodal_adatas({"RNA": source_adata_RNA, "ATAC": source_adata_ATAC})
target_adata = sd.utils.combine_multimodal_adatas({"RNA": target_adata_RNA, "ATAC": target_adata_ATAC})
# 4) Spatial graphs
source_edge_index = sd.utils.get_multi_edge_index(source_adata.obsm["spatial"], source_adata.obs["batch"], n_neighbors=10)
target_edge_index = sd.utils.get_single_edge_index(target_adata.obsm["spatial"], n_neighbors=10)
# 5) Initialize DIRAC annotation app
annotate = sd.main.annotate_app(save_path=save_path, use_gpu=True)
# 6) Define modality splits in the concatenated matrix
# RNA: [0, dim_RNA)
# ATAC: [dim_RNA, dim_RNA + dim_ATAC)
split_list = [
(0, source_adata_RNA.shape[1]),
(source_adata_RNA.shape[1], source_adata_RNA.shape[1] + source_adata_ATAC.shape[1]),
]
# 7) Package data (note split_list)
samples = annotate._get_data(
source_data=source_adata.X,
source_label=source_adata.obs["cell.type"],
source_edge_index=source_edge_index,
target_data=target_adata.X,
target_edge_index=target_edge_index,
num_parts_source=source_adata.shape[0] // 200,
num_parts_target=target_adata.shape[0] // 200,
split_list=split_list,
)
# 8) Build model and train
models = annotate._get_model(samples=samples, opt_GNN="SAGE")
results = annotate._train_dirac_annotate(samples=samples, models=models, n_epochs=100)
# 9) Write back outputs
source_adata.obsm[f"{methods}_embed"] = results["source_feat"]
target_adata.obsm[f"{methods}_embed"] = results["target_feat"]
target_adata.obs[f"{methods}_pred"] = results["target_pred"]
target_adata.obs[f"{methods}"] = target_adata.obs[f"{methods}_pred"].map(results["pairs"])
# 10) Metrics & Visualization
metrics_all = {
"Accuracy Score":
float(sklearn.metrics.accuracy_score(target_adata.obs["cell.type"], target_adata.obs[f"{methods}"])),
"Precision Score":
float(sklearn.metrics.precision_score(target_adata.obs["cell.type"], target_adata.obs[f"{methods}"], average='weighted')),
"Recall Score":
float(sklearn.metrics.recall_score(target_adata.obs["cell.type"], target_adata.obs[f"{methods}"], average='weighted')),
"F1 Score":
float(sklearn.metrics.f1_score(target_adata.obs["cell.type"], target_adata.obs[f"{methods}"], average='weighted'))}
print(metrics_all)
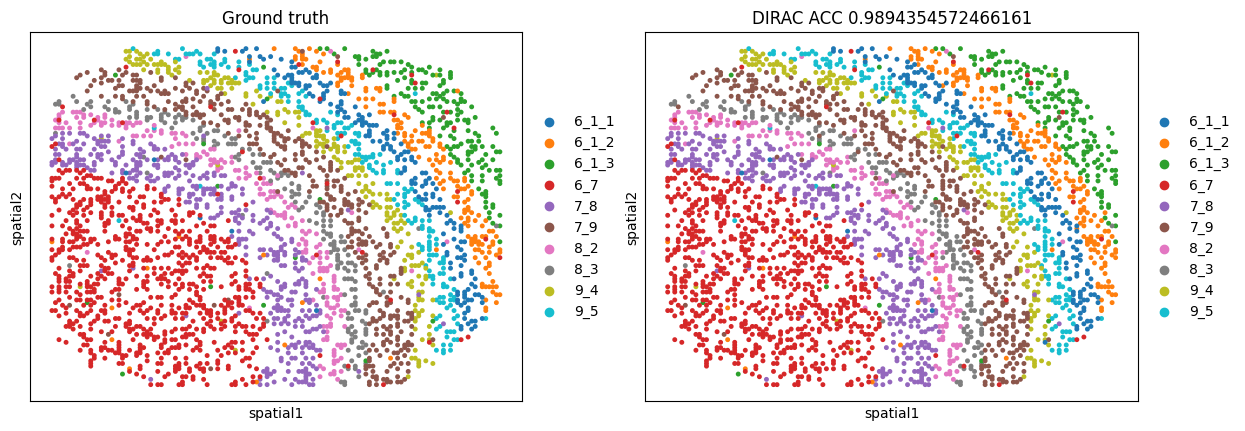
sc.pl.embedding(target_adata, basis='spatial', color=['cell.type', f'{methods}'], title=['Ground truth',f"{methods} ACC {metrics_all['Accuracy Score']}"], s=50, show=False)
AnnData object with n_obs × n_vars = 11971 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 3029 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 11971 × 600
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 3029 × 600
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
Average neighbors per node (directed): 10.00 (edges=119710, nodes=11971)
Average neighbors per node (directed): 10.00 (edges=30290, nodes=3029)
Identified 2 unique domains.
Computing METIS partitioning...
Done!
Computing METIS partitioning...
Done!
DIRAC annotate training..: 100%|█| 10
{'Accuracy Score': 0.9894354572466161, 'Precision Score': 0.9894781692014127, 'Recall Score': 0.9894354572466161, 'F1 Score': 0.9894421771682003}
[4]:
[<Axes: title={'center': 'Ground truth'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'DIRAC ACC 0.9894354572466161'}, xlabel='spatial1', ylabel='spatial2'>]
7. Novel Cell Type Discovery (Confidence Filtering)¶
Why this matters. Real references are often incomplete: some cell types may be absent. Here we intentionally drop two cell types from the reference, train DIRAC, and then use a confidence threshold (default 0.9, tunable) to mark low-confidence predictions as ``”unassigned”``. This flags putative novel/unknown or out-of-reference cell types in the target.
What happens.
Randomly remove two reference cell types (
'7_9','6_1_2'in this demo).Train DIRAC on the pruned reference and full target (RNA+ATAC).
Use
filter_low_confidence=Truewithconfidence_threshold=0.9.Post-process: points below the threshold become
"unassigned".Report metrics on the remaining assigned cells and visualize confidence.
Tip. Increase the threshold (e.g., 0.95) for stricter assignment; relax it (e.g., 0.8) on noisier data.
[5]:
# 1) Load inputs
# RNA
source_adata_RNA = anndata.read_h5ad(os.path.join(data_path, "source_RNA.h5ad"))
target_adata_RNA = anndata.read_h5ad(os.path.join(data_path, "target_RNA.h5ad"))
print(source_adata_RNA); print(target_adata_RNA)
# ATAC
source_adata_ATAC = anndata.read_h5ad(os.path.join(data_path, "source_ATAC.h5ad"))
target_adata_ATAC = anndata.read_h5ad(os.path.join(data_path, "target_ATAC.h5ad"))
print(source_adata_ATAC); print(target_adata_ATAC)
####### remove some cell types from reference data
drop_celltypes = ['7_9', '6_1_2']
source_adata_RNA = source_adata_RNA[~source_adata_RNA.obs['cell.type'].isin(drop_celltypes)].copy()
source_adata_ATAC = source_adata_ATAC[~source_adata_ATAC.obs['cell.type'].isin(drop_celltypes)].copy()
# 2) Preprocess each modality
sc.pp.normalize_total(source_adata_RNA); sc.pp.log1p(source_adata_RNA); sc.pp.scale(source_adata_RNA)
sc.pp.normalize_total(target_adata_RNA); sc.pp.log1p(target_adata_RNA); sc.pp.scale(target_adata_RNA)
sc.pp.normalize_total(source_adata_ATAC); sc.pp.log1p(source_adata_ATAC); sc.pp.scale(source_adata_ATAC)
sc.pp.normalize_total(target_adata_ATAC); sc.pp.log1p(target_adata_ATAC); sc.pp.scale(target_adata_ATAC)
# 3) Concatenate modalities (feature-wise)
source_adata = sd.utils.combine_multimodal_adatas({"RNA": source_adata_RNA, "ATAC": source_adata_ATAC})
target_adata = sd.utils.combine_multimodal_adatas({"RNA": target_adata_RNA, "ATAC": target_adata_ATAC})
# 4) Spatial graphs
source_edge_index = sd.utils.get_multi_edge_index(source_adata.obsm["spatial"], source_adata.obs["batch"], n_neighbors=10)
target_edge_index = sd.utils.get_single_edge_index(target_adata.obsm["spatial"], n_neighbors=10)
# 5) Initialize DIRAC annotation app
annotate = sd.main.annotate_app(save_path=save_path, use_gpu=True)
# 6) Define modality splits in the concatenated matrix
# RNA: [0, dim_RNA)
# ATAC: [dim_RNA, dim_RNA + dim_ATAC)
split_list = [
(0, source_adata_RNA.shape[1]),
(source_adata_RNA.shape[1], source_adata_RNA.shape[1] + source_adata_ATAC.shape[1]),
]
# 7) Package data (note split_list)
samples = annotate._get_data(
source_data=source_adata.X,
source_label=source_adata.obs["cell.type"],
source_edge_index=source_edge_index,
target_data=target_adata.X,
target_edge_index=target_edge_index,
num_parts_source=source_adata.shape[0] // 200,
num_parts_target=target_adata.shape[0] // 200,
split_list=split_list,
)
# 8) Build model and train
models = annotate._get_model(samples=samples, opt_GNN="SAGE")
results = annotate._train_dirac_annotate(samples=samples, models=models, n_epochs=100,
filter_low_confidence = True, # enable filtering
confidence_threshold = 0.9) # tune this (e.g., 0.8–0.95)
# 9) Write back outputs
target_adata.obs[f"{methods}_pred"] = results["target_pred_filtered"]
target_adata.obs[f"{methods}"] = target_adata.obs[f"{methods}_pred"].map(results['pairs_filter'])
target_adata.obs["Confidence"] = results['target_confs']
# 10) Metrics (on assigned cells only) & Visualization
valid_mask = target_adata.obs[f"{methods}"] != "unassigned"
filtered_target = target_adata[valid_mask].copy()
metrics_all = {
"Accuracy Score": float(sklearn.metrics.accuracy_score(
filtered_target.obs["cell.type"],
filtered_target.obs[f"{methods}"])),
"Precision Score": float(sklearn.metrics.precision_score(
filtered_target.obs["cell.type"],
filtered_target.obs[f"{methods}"],
average='weighted', zero_division=0)),
"Recall Score": float(sklearn.metrics.recall_score(
filtered_target.obs["cell.type"],
filtered_target.obs[f"{methods}"],
average='weighted')),
"F1 Score": float(sklearn.metrics.f1_score(
filtered_target.obs["cell.type"],
filtered_target.obs[f"{methods}"],
average='weighted')),
"Unassigned Rate": float(1 - len(filtered_target)/len(target_adata))
}
sc.pl.embedding(target_adata, basis='spatial', color=['cell.type', f'{methods}', "Confidence"],
title=['Ground truth',f"DIRAC ACC {metrics_all['Accuracy Score']}", "Confidence"],
s=80, show=False, color_map="CMRmap")
AnnData object with n_obs × n_vars = 11971 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 3029 × 200
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 11971 × 600
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
AnnData object with n_obs × n_vars = 3029 × 600
obs: 'pop', 'depth', 'cell.type', 'cell.type.idx', 'batch'
obsm: 'spatial'
Computing METIS partitioning...
Done!
Computing METIS partitioning...
Done!
Average neighbors per node (directed): 10.00 (edges=95610, nodes=9561)
Average neighbors per node (directed): 10.00 (edges=30290, nodes=3029)
Identified 2 unique domains.
DIRAC annotate training..: 100%|█| 10
[5]:
[<Axes: title={'center': 'Ground truth'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'DIRAC ACC 0.9123914759273876'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'Confidence'}, xlabel='spatial1', ylabel='spatial2'>]

Summary
Single-modality (RNA→RNA): build source/target spatial graphs → train with GraphSAGE → predict → metrics → spatial maps.
Dual-modality (RNA+ATAC): concatenate features and provide
split_list; rest is identical.Novel cell type discovery: train with confidence filtering to mark “unassigned” cells when the reference is incomplete; report Unassigned Rate alongside standard metrics.
Use
num_parts_*to trade memory for speed, and tune neighbors/model/thresholds for stable, well-mixed embeddings and reliable annotations.